

Is digital marketing in MedTech and Pharma similar? Understanding the specifics of MedTech and Digital Health digital marketing is essential to success.
MedTech digital marketing
The four main components of digital marketing in the medical devices industry are digital strategy consulting, digital content creation and management, digital campaign execution, and analytics.
- Digital Marketing Consulting involves helping MedTech companies assess their priorities and strengths, optimize their operations and innovation, and develop effective go-to-market strategies.
- Digital content creation and management involves producing engaging and informative content for various channels such as email, social media, blogs, webinars, etc. that showcase medical devices’ value proposition and benefits.
- Digital campaign execution involves designing and implementing marketing campaigns that target specific segments of healthcare professionals (HCPs) or patients using digital tools such as CRM systems, marketing automation platforms, SEO/SEM techniques, etc.
- Digital analytics involves measuring and evaluating the performance and impact of digital marketing activities using data-driven methods such as web analytics, customer feedback surveys, ROI calculations, etc.
Digital consulting is a vital component of digital marketing for medical devices and should be a first step. It helps MedTech companies to:
- Define their vision and goals for digital transformation
- Assess their current capabilities and gaps
- Develop a roadmap and action plan for implementing digital solutions
- Align their organization and culture with digital best practices
Digital marketing of medical devices in comparison to pharmaceuticals
Digital marketing for Medical Devices in many aspects is similar to the pharmaceutical industry, however, there are significant differences.
What are the similarities between MedTech and Pharma digital marketing?
- Both Pharma and MedTech use digital tools to inform and influence HCPs and patients about their products and services.
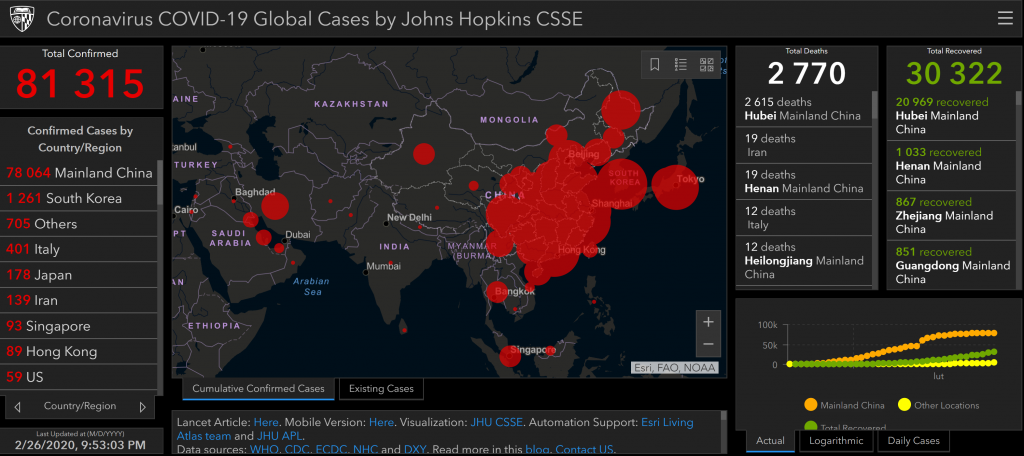
- Both industries rely on data and analytics to measure and optimize their digital marketing activities.
- Medical Devices face regulatory challenges and compliance issues when engaging with their audiences online that are similar to those in pharmaceuticals.
The key differences between Pharma and Medical Devices digital marketing are that:
- Medtech companies tend to have more complex products that require more technical expertise and demonstration than pharma companies.
- Medtech companies have a wider range of stakeholders to consider, such as hospital administrators, payers, distributors, etc. than pharma companies.
- Medtech companies have more opportunities to leverage digital health and digital therapeutics (DTx), such as connected devices, apps, sensors, etc. that can enhance their value proposition and customer experience than pharma companies
Outsourcing digital marketing services in the MedTech industry
Medical device companies may look for different services or support when deciding to outsource digital marketing, depending on their needs and goals.
Some possible services or support are:
- Quality assurance: ensuring that the digital marketing activities comply with regulatory standards and best practices.
- Content production and management: producing digital content or products such as websites, apps, videos, etc. that showcase the features and benefits of medical devices.
- E-commerce: setting up and managing online platforms that allow customers to order and purchase medical devices easily and securely.
- Virtual sales channels: creating and maintaining digital tools that enable sales reps to communicate and demonstrate medical devices to HCPs remotely.
- Data analytics: collecting and analyzing data from digital marketing activities to measure performance, optimize campaigns, and generate insights
Generally, smaller MedTech companies may need more comprehensive and flexible services and support than larger companies, as they may have less experience and capacity for digital marketing. Larger companies may need more specialized and customized support than smaller companies, as they may have more complex and diverse needs for digital marketing.
Some possible factors that influence digital marketing service requirements are:
- The type and complexity of medical devices that the company produces or sells.
- The level of expertise and resources that the company has internally for digital marketing.
- The scope and scale of digital marketing activities that the company wants to undertake.
- The budget and timeline that the company has for digital marketing outsourcing.
The skillset of MedTech digital marketer
Some of the skills of a digital marketing expert from pharma are transferrable to provide medical device digital marketing services too. For example, both sectors require:
- Knowledge of regulatory requirements and compliance standards.
- Ability to create engaging and informative content for different audiences and channels.
- Proficiency in using various digital tools and platforms to design, execute, and measure campaigns.
However, there are also some differences between pharma and medical device digital marketing that may require additional skills or adaptation. The key differences are that:
- Medical device customers have different expectations and needs than pharma customers. They may be more interested in product features, benefits, demonstrations, or testimonials than in disease mechanisms or outcomes.
- Medical devices are more diverse and complex than drugs. They can range from tissue grafts to prostheses to digital devices and apps. They may also have different modes of action, indications, or usage scenarios.
- Medical devices may have shorter product life cycles than drugs. They may face more competition or innovation from other players in the market. They may also require more frequent updates or upgrades.
Therefore, a digital marketing expert to excel in the Medical Devices industry may need to understand the nuances of device marketing and how to tailor their strategies accordingly. MedTech digital marketing specialist has to learn about the specific types of devices they are marketing and how they work, who they serve, and what value they offer. Finally, digital marketer in MedTech industry has to be flexible and agile in responding to changing market conditions and customer feedback.
Regulatory requirements for medical device digital marketing
Regulatory requirements for medical device digital marketing vary depending on the type of device, the market, and the channel. Some of the general requirements are:
- Medical device digital marketing must be truthful, accurate, and not misleading.
- Medical device digital marketing must comply with the relevant laws and regulations of each country or region where they operate.
- Medical device digital marketing must respect the data privacy and security of customers and users.
Some examples of specific regulatory requirements for medical device digital marketing are:
- The Medical Devices Regulation (MDR) applies since 26 May 2021 in the European Union. It sets out new rules for placing medical devices on the market, including requirements for clinical evidence, post-market surveillance, labeling, and advertising.
- The In Vitro Diagnostic Devices Regulation (IVDR) applies since 26 May 2022 in the European Union. It replaces Directive 98/79/EC and introduces new classification rules, conformity assessment procedures, performance evaluation requirements, etc. for in vitro diagnostic devices.
- The Food and Drug Administration (FDA) provides guidance documents with digital health content for medical device manufacturers in the United States. They cover topics such as software as a medical device, mobile medical applications, clinical decision support software, cybersecurity, artificial intelligence/machine learning, etc.
Read our Expert Guide for 2025: Medical Device Digital Marketing













{kind=link}