
The EU AI Act Deadline Moved. The Commercial Operating Model Cannot Wait.
The EU AI Act has given pharma, MedTech and digital health companies more time on the most resource-intensive part of the regime.
It has not given commercial teams permission to wait.
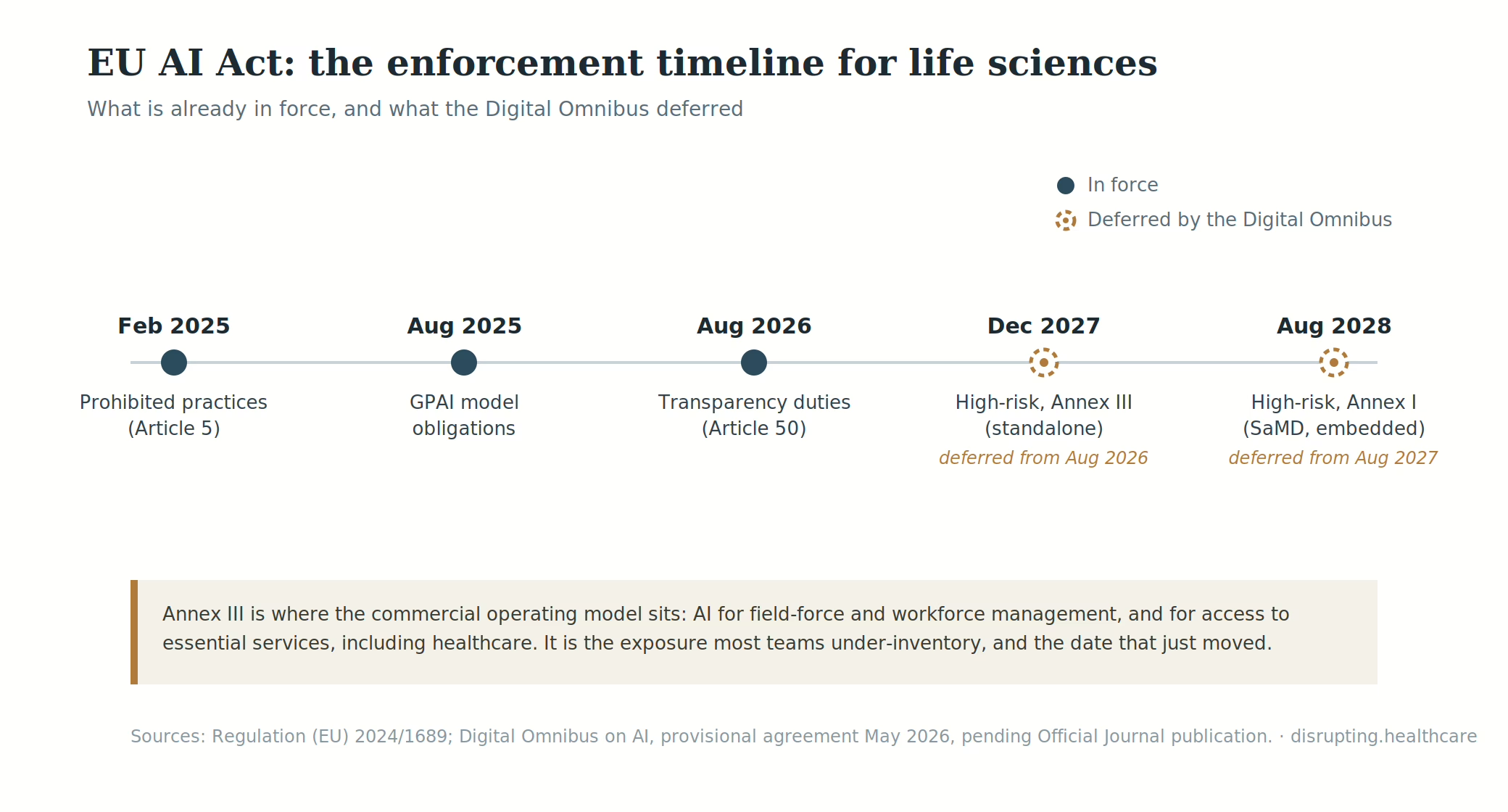
On 29 June 2026, the Council of the European Union gave final approval to the Digital Omnibus on AI. The package moves the high-risk obligations to 2 December 2027 for systems classified through Annex III, and to 2 August 2028 for high-risk AI connected to products covered by EU sectoral legislation, including qualifying medical devices and IVDs.
The legislative act still needs publication in the Official Journal and will enter into force on the third day after publication. But the political and regulatory direction is now clear: for many AI-enabled medical devices, clinical decision-support tools and software products, the formal compliance runway has lengthened.
That is the headline. It is not the real story.
The real story is that AI governance in life sciences is no longer a legal workstream sitting somewhere between privacy, regulatory affairs and IT. It is becoming part of the commercial operating model: how companies select vendors, design HCP engagement, run patient support, generate evidence, manage content and decide which tools can safely move from pilot to production.
For commercial leaders, the risk is not only regulatory non-compliance. It is building an AI-enabled commercial model that cannot survive procurement scrutiny, MLR review, market-access due diligence or hospital trust checks.
Lovely PowerPoint, terrible aftertaste.
What changed in the EU AI Act timeline—and what did not?
The Digital Omnibus changes the timetable for high-risk systems. It does not reset the rest of the AI Act.
The Act entered into force on 1 August 2024. Prohibited AI practices and AI-literacy obligations have applied since February 2025, while governance rules and obligations for general-purpose AI models began applying in August 2025. Much of the remaining regulation becomes applicable from 2 August 2026.
Article 50 transparency obligations also apply from 2 August 2026. They cover areas such as informing people when they are interacting directly with an AI system and marking or labelling certain AI-generated or manipulated content.
There is a narrower transition until 2 December 2026 for providers of systems that generate synthetic audio, images, video or text and were already on the market before 2 August 2026. Under the Council-approved text, that transition applies specifically to Article 50(2). It is not a general postponement of every transparency obligation.
For commercial teams, the practical sequence is therefore straightforward. AI-literacy and prohibited-practice controls should already exist. Customer-facing transparency work belongs in the 2026 plan. Annex III high-risk obligations move to December 2027. Qualifying product-related obligations move to August 2028.
The dates have changed. The need to know what your systems actually do has not.
Two high-risk routes, two different clocks
The distinction between the 2027 and 2028 deadlines matters because many life sciences companies are watching the wrong clock.
The first route covers systems classified as high-risk under Article 6(2) and Annex III. Annex III covers specified applications in areas including employment, worker management, biometrics and access to certain essential services.
For commercial organisations, the most immediate exposure may sit in workforce technology. An AI system that materially influences recruitment, performance assessment, promotion, work allocation or termination may require a serious Annex III review.
That does not make every sales dashboard, territory tool or field-force recommendation engine automatically high-risk. Article 6 allows some Annex III systems to fall outside the high-risk category where they perform narrow, preparatory or procedural tasks and do not materially influence a decision. The assessment still needs to be documented.
Patient-access and eligibility tools require similar precision. Annex III covers systems used by, or on behalf of, public authorities to assess eligibility for public assistance benefits and services, including healthcare. A patient-support chatbot operated by a pharmaceutical company is not automatically high-risk simply because it discusses access. A system influencing entitlement to a covered healthcare service may be another matter.
The second route covers AI that is itself a product, or a safety component of one, covered by legislation listed in Annex I and subject to third-party conformity assessment.
This is the route most relevant to many AI-enabled medical devices, IVDs, clinical decision-support products and connected diagnostic systems. It is now associated with the 2 August 2028 deadline.
Again, not every medical application of AI qualifies. Article 6 requires both a connection to a product covered by Annex I legislation and a third-party conformity assessment. The product’s intended purpose and classification under the MDR or IVDR remain decisive.
A product team developing AI-enabled software as a medical device may therefore be working towards the 2028 route. A commercial organisation deploying AI to evaluate employees may be closer to 2027. An HCP segmentation model may fall under neither high-risk route, while still raising important questions under GDPR, pharmaceutical promotion rules, security policies and internal AI governance.
The Act does not care whether the system sits in a product roadmap or the commercial technology stack.
It cares what the system does.
Why commercial teams are watching the wrong systems
The easiest mistake is to treat the EU AI Act as another regulatory checklist focused on products.
That is too small.
AI is moving into precisely the places where commercial judgement, medical governance, evidence, data and customer trust meet. It already supports segmentation, next-best-action recommendations, content adaptation, payer analytics, forecasting, field-force planning, patient navigation and digital product development.
Some of these uses will be high-risk. Many will not. Almost all will require clearer ownership than the average innovation pilot currently enjoys.
Vendor selection is the first pressure point. Most commercial AI capability is not built entirely in-house. It arrives through CRM extensions, analytics platforms, agency tools, medical-information systems, content engines, patient-support vendors and field-force productivity products.
This creates a basic operating risk: the company may buy a tool before it understands the intended purpose, classification, data flows, limitations or evidence available from the provider. Commercial procurement needs to establish what the system influences, which legal role each party holds, how updates are controlled, what can be audited and what happens when the model changes.
“We use responsible AI” is a positioning statement. It is not a vendor file.
Launch planning is the second. An AI-enabled device, companion application or remote-monitoring product can clear one regulatory gate and still struggle commercially if the company has not modelled the AI Act, MDR or IVDR, GDPR, cybersecurity and evidence requirements together.
The regulatory work shapes the commercial proposition. It affects what can be claimed, which evidence must be generated, how the product is implemented and what hospitals or procurement teams can reasonably ask to see. AI governance therefore belongs in launch sequencing, not in a legal annex added after the value dossier is complete.
Market access is the third. Clinical evidence remains the centre of the argument. But weak model governance can make otherwise credible evidence harder to trust. Payers and hospital procurement teams do not need another lecture about the transformative power of AI. They need to understand how the product performs, how it fits into workflow, how changes are controlled and who is accountable when the output is wrong.
MLR and content operations are the fourth. Generative AI can produce more content than a regulated organisation can safely review. That is not necessarily innovation. Sometimes it is simply a compliance treadmill with better shoes.
When AI enters promotional adaptation, disease-awareness content, medical education or HCP support, the organisation needs provenance, approved source controls, version history, prompt governance and defined human review. The key question is not whether AI helped to produce the material. It is whether the company can reconstruct how the final claim reached the customer: which system was used, which sources were available, what a human changed and who approved publication.
Article 50 adds a separate transparency layer for certain AI-generated content and direct interactions with AI systems. Public disclosure and internal MLR provenance are not the same obligation, but commercial teams need a design for both.
Customer-facing interfaces make the issue visible. Under Article 50, people generally need to be informed when they are interacting directly with an AI system unless that is obvious from the circumstances. For HCP portals, patient-support services and AI-enabled medical-information tools, disclosure should be part of the experience rather than buried in terms and conditions.
The harder questions follow immediately. What can the interface answer? When must it escalate to a human? Which sources can it use? How is a misleading response detected? Who owns the interaction after the vendor’s demo team has left the building?
Transparency is not a footer. It is part of the service design.
This is the operating-model issue explored in Agentic AI in Life Sciences Commercial: What Actually Changes in Your Operating Model? Accountability must follow the decision, not disappear into the technology stack.
A CE mark does not close the AI Act question
For AI-enabled medical devices, the CE mark remains essential. It is not the whole answer.
Joint guidance from the Artificial Intelligence Board and Medical Device Coordination Group makes clear that the MDR or IVDR and the AI Act can apply to the same product. It also explains how medical-device classification and the applicable conformity-assessment route help determine whether an AI system meets the Article 6(1) high-risk conditions.
The important point is not that manufacturers should create two entirely separate compliance bureaucracies. Existing product documentation, quality systems and conformity-assessment processes can be used to address overlapping requirements where appropriate.
The Digital Omnibus also introduces mechanisms intended to reduce duplication where sectoral legislation already contains similar AI-specific requirements. Overlap can therefore be managed. It should not be confused with exemption.
For launch and market-access teams, the practical question is sequencing. Which evidence will support both regulatory conformity and adoption? Which model changes could affect claims or performance? What documentation will a notified body, hospital, payer or procurement team expect? Who will explain the system’s limitations to the customer?
The answer needs to be built into the commercialisation model. As discussed in Digital Health Commercialisation in Europe, regulatory permission is only one part of adoption. Procurement, workflow integration, evidence and local execution remain stubbornly important.
A CE mark opens the regulatory door.
Increasingly, the governance file will help the product through procurement.
The operating question: who owns the decision?
The AI Act defines responsibilities for providers, deployers and other operators.
Commercial organisations still need to translate those legal roles into actual decision rights.
Who owns classification? When a country team proposes a new AI-enabled HCP engagement tool, who decides whether it is high-risk, subject to transparency requirements or ordinary enterprise software?
Legal cannot answer that alone. Neither can IT. The decision requires commercial context, regulatory interpretation, data protection, procurement and, depending on the use case, medical or product governance.
Who owns the output? If a system recommends that one HCP should receive priority over another, or that a patient should enter a particular support pathway, who is accountable for acting on that recommendation?
The vendor may have built the model. It does not follow that the vendor owns the business decision.
Who owns monitoring? Models drift, input data changes, products evolve and local teams find creative new uses for tools that were approved for something narrower.
Governance needs thresholds, review intervals, escalation rules and evidence that somebody is looking at performance after launch—not merely celebrating deployment.
And who owns the stop button? Human oversight is not a slogan. It requires the authority to override or suspend a system when performance, bias, safety or compliance concerns emerge.
This is where elegant AI strategy starts sweating through its shirt.
Europe has granted runway, not absolution
The revised timeline creates room to build the operating model properly.
Squandering that room would be impressively familiar, but still a bad idea.
The first move is a commercial AI register covering systems used across CRM, content, analytics, market access, patient support, field operations and regulated digital products. The useful fields are not merely the technology name and vendor. They are intended purpose, decisions influenced, users affected, markets, legal role, classification view, human oversight and named owner.
Classification should then become part of the production gate. A pilot should not enter routine commercial use because it performed well in a controlled demonstration. The intended use, data flows, vendor evidence and operating controls should be reviewed before scale-up—and again when a market changes the use case.
Vendors should provide a usable evidence file covering intended purpose, known limitations, update policy, auditability, security controls and model dependencies. Contracts should allocate responsibility for documentation, incidents and material changes rather than assuming the answer will become obvious during a crisis.
AI governance also needs to connect with MLR and content operations. The same model may support internal training, medical education, and promotional adaptation, but those uses should not share the same approval pathway. Governance should follow the use case and its consequences.
Finally, monitoring should become part of commercial performance management. The dashboard should not stop at adoption, activity, or ROI. It should also show overrides, complaints, error patterns, output quality, model changes and drift from the approved purpose.
The Digital Omnibus is helpful. It gives companies more time to address standards, documentation, conformity, and the practical overlap between the AI Act and sectoral regulation.
The strongest companies will use that time to build a repeatable operating model across commercial, medical, legal, regulatory, data, security and procurement.
The weaker response will be to treat 2027 and 2028 as distant compliance dates while AI pilots continue multiplying without clear ownership.
That would miss the strategic point.
The winners will not be the companies with the largest collection of AI tools. They will be the ones that know which decisions those tools influence, who owns the outcome, what evidence supports their use and when the system should be stopped.
The high-risk deadline moved.
Accountability did not.
This article provides strategic analysis and does not constitute legal advice.